This is an old revision of the document!
The CAMDA Challenges
As traditional in CAMDA contests, neither we nor the producers of the data can provide advice on the datasets to individuals as dealing with the files forms part of the analysis challenge. There is, however, an open forum for participants' free discussions relating to the contest data sets, and in which you are encouraged to participate.
We look forward to a lively contest!
Dataset 1: TGP dataset from the Japanese Toxicogenomics Project
The TGP dataset contains over >21,000 arrays for rats treated with mainly human drugs and profiled using the Affymetrix RAE230_2.0 GeneChip®. The main target organ profiled is liver.
In this project, only the data for liver are provided. The data package contains the following files:
- TGP Description (word document) – it provides a brief introduction of the TGP data and human hepatotoxic potential of each drug. More information is available from two references below: Citation 1: Uehara T, Ono A, Maruyama T, Kato I, Yamada H, Ohno Y, Urushidani T., The Japanese toxicogenomics project: application of toxicogenomics. Mol Nutr Food Res. 54(2):218-27, 2010. Citation 2: Chen, M., et al., FDA-approved drug labeling for the study of drug-induced liver injury (DILI). Drug Discov Today, 2011. 16(15-16): p. 697-703.
- Drug Information (Excel table) – the basic information about individual drugs are extracted from DrugBank. The last three columns contain human hepatotoxicity data for each drug described in the paper by Chen et al. (mentioned above in citation 1).
- Pathology Data (Excel table) – A significant portion of the TGP data is derived from in vivo assay using two different treatment protocols (i.e., single treatment and daily repeated treatment). Pathology and clinical chemistry data for each rat (which anchored with each array) are summarized in this table.
- Array Metadata (csv format) – Meta data (e.g., dose, time, sacrifice time and etc) for each array are summarized. Phenotypic data anchored to each array are available from the “Pathology data” table mentioned above.
- MAS5 data (folder) – it contains the MAS5 summarized array data
- FARMS data (csv format) – contain the FARMS summarized array data
- RAW data (folder) – it contains all the array data in the cel format
- Example data (LIBSVM format) - ready to use for binary classification of DILI
Challenges
This is a typical toxicogenomics dataset. This dataset can be used to address two most important questions in toxicology and safety evaluation:
- Question 1: Can we replace the animal study with in vitro assay? The current safety assessment is largely relied on the animal model, which is time-consuming, labor-intensive, and definitely not in line with the animal right voice. There is a paradigm shift in toxicology to explore the possibility of replacing the animal model with in vitro assay coupled with toxicogenomics. The TGP data contains both in vitro and animal data, which is essential to address this question.
- Question 2: Can we predict the liver injury in humans using toxicogenomics data from animals. Around 40% of drug-induced liver injury (DILI) cases are not detected in the preclinical studies using the conventional indicators (such as pathology, clinical chemistry data). It has been hypothesized that genomic biomarkers will be more sensitive than conventional markers in detecting human hepatotoxicity signals in preclinical studies (i.e., in vitro and in vivo assays). In this project, we provide the human hepatotoxicity data for most of the drugs (the last three columns in the table named “Drug Information”). The contests can explore the possibility of predicting the DILI potential in humans using the in vitro data from rat primary hepatocytes or human primary hepatocytes, or the animal data from two different treatment protocols. Alternatively, these data can also be combined to enhance the predictive power for the human hepatotoxic potential.
Please notice, the CAMDA challenge is not limited to these questions!
Data download Preprocessed data (FARMS summarized) is available in several formats (CSV, LIBSVM) and can be downloaded here. Participants who want to use the raw data (CEL-files) or the MAS5.0 summarized expression values should read and accept the data download agreement to get access.
Dataset 2: KPGP-38 Human Genomes
Both bioinformatics and medical informatics professionals are challenged with massive data generated by genome sequencing and complex data from the electronic medical records. How do we leverage these data to improve patient care? The second track of CAMDA will address some of the issues. The purpose of this track is exploratory. We see it more of a collaborative effort from the community to define the problems, rather than a competition among groups for the best answer. Thus, tasks in this track are less defined, and regular conference calls will be held for discussions.
Data Description
38 Human genomes sequenced on Illumina HiSeq 2000 platform with 30x to 40x coverage. Human subjects are part of the Korean Personal Genome Project, which is part of the international Personal Genome Project (PGP). Limited medical record data is available for the subjects.
Challenges
We invited innovative ideas on utilizing the data set for developing novel approaches to analyzing big data. Some interesting questions might be:
- Question 1: How can single nucleotide variants (SNVs) be called and how can they be distinguished from systematic sequencing errors?
Variant calling:
One of the first tasks when analyzing sequencing data is base calling and, in particular, variant calling. The KPGP data has pedigree information which allows verifying variant calling because the vast majority of variants are inherited from the parents.
Sequencing errors:
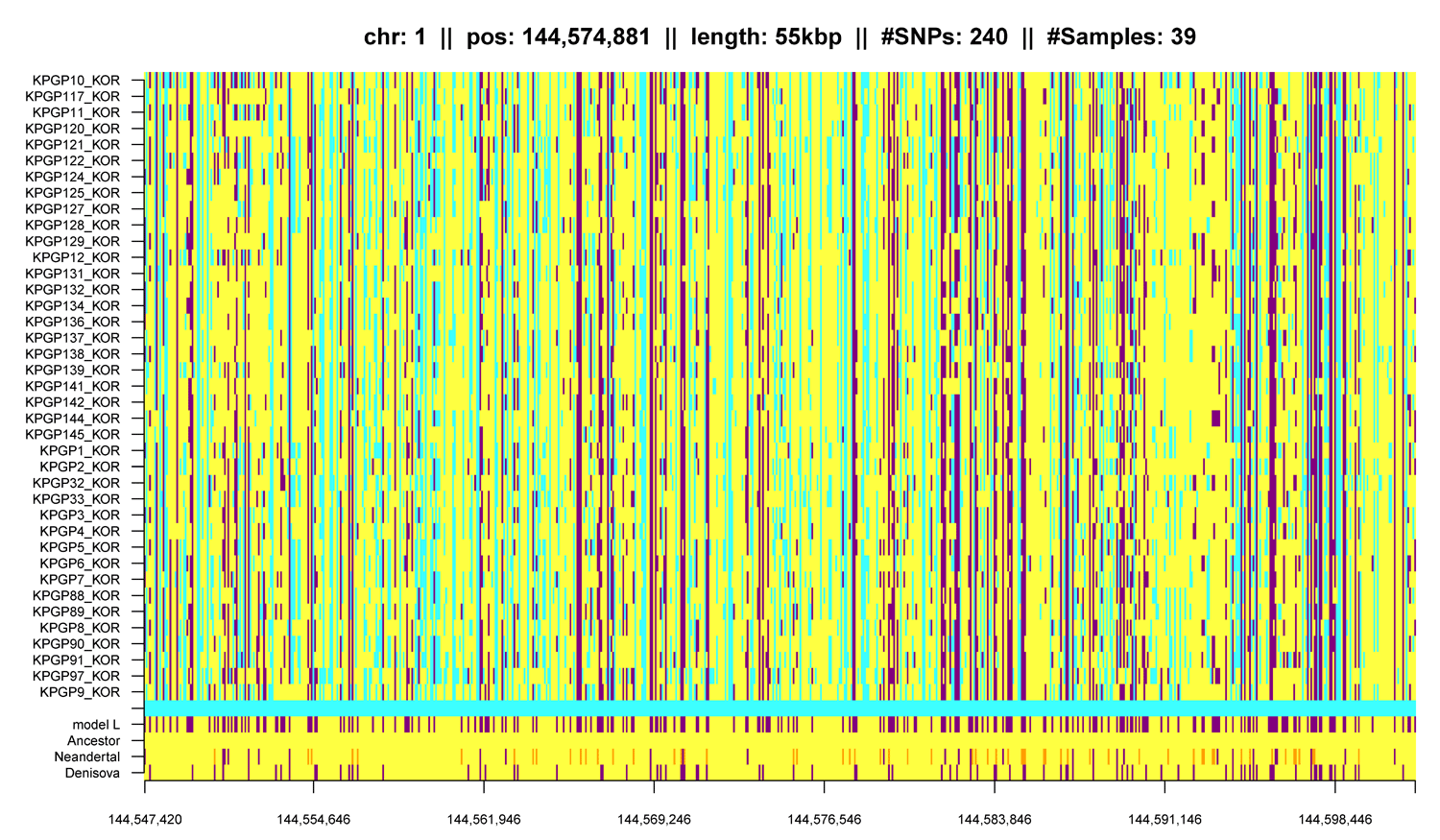
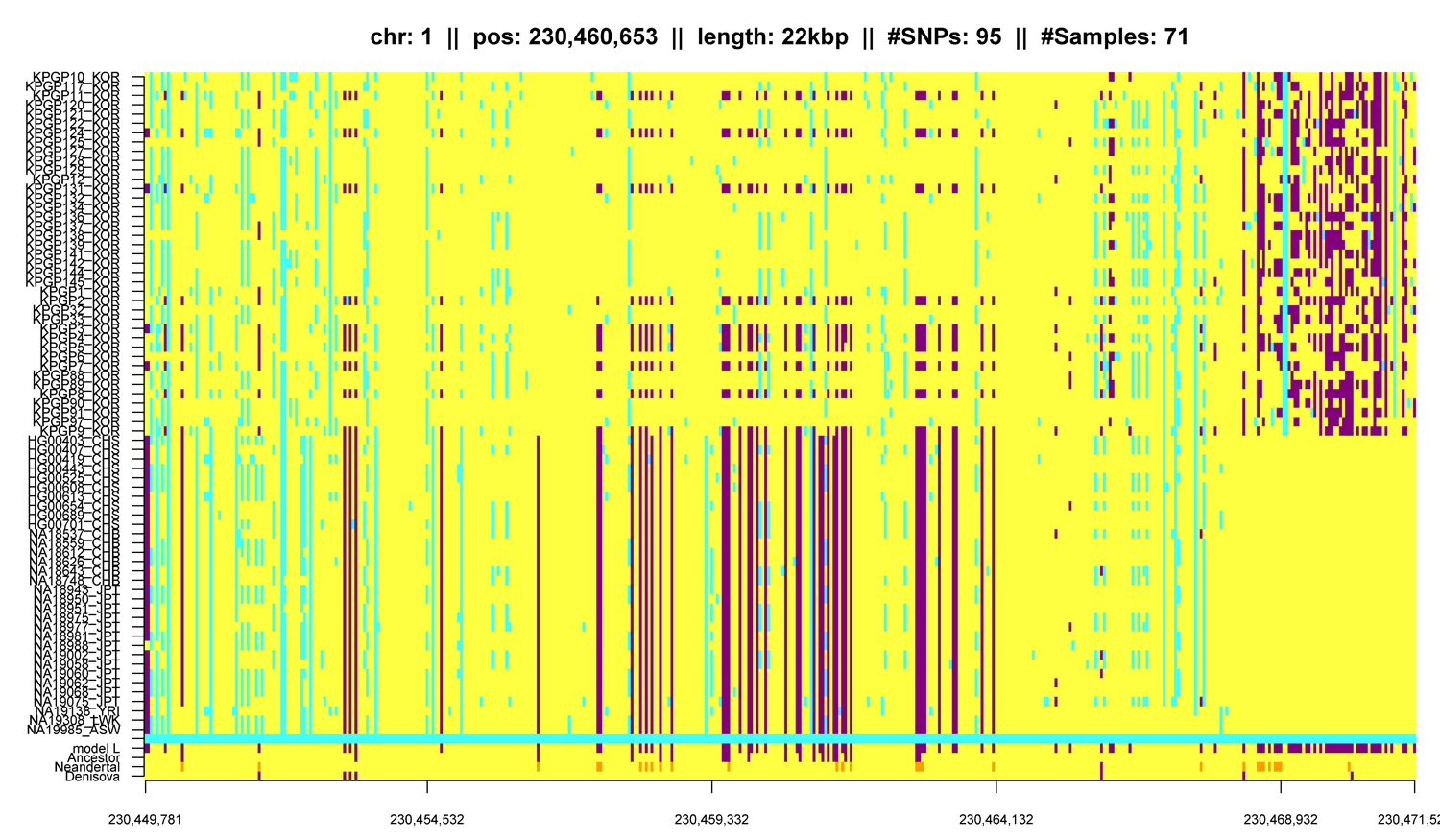
Variant calling must be distinguished from systematic sequencing errors as found in the KPGP data (see Hochreiter). Systematic sequencing errors can be detected in two twin pairs (KPGP88/KPGP89 and KPGP90/KPGP91), or in one KPGP individual that is an Caucasian female. Variants that appear only in Koreans should not be present in this Caucasian female - otherwise the variants would be characteristic for the sequencing center or the data preprocessing. For this analysis data like the 1000 Genomes Project data may be used to verify whether the variants appear only in Koreans. Further the KPGP data contains regions where variants accumulate which may hint at sequencing errors. - Question 2: Can structural variants be reliably called?
Structural variants like deletions, duplications, inversions, translocations, or copy number variants are important in analyzing sequencing data. Copy number variants and other structural variants can be verified using the pedigree information. Most structural variants are inherited and, therefore, are present in both the child and one of the parents. In a further step, the detection of de novo structural variants is of interest. However the evaluation of the detection accuracy may be more difficult as false discoveries are hard to identify. - Question 3: Can segments of identity by descent (IBD) be found?
Two genomes are identical by descent (IBD) if they share a DNA segment that both inherited from a common ancestor. Can IBD segments be found? How reliable can they be found? How short are they? For this task again the pedigree information is available to verify whether IBD segments are shared by related individuals. These IBD segments are assumed to be long and more easily to be detected. A more challenging task is to find IBD sharing between unrelated individuals. - Question 4: How is the Korean genome related to other genomes?
The idea of IBD sharing can be extended to other population, e.g. by including data from the 1000 Genomes Project. Using IBD or SNV sharing, the degree of matches with genomes from other populations can be assessed. Other populations might be Africans or Europeans but also Japanese, Han Chinese from Beijing or Han Chinese from South as provided by the 1000 Genomes Project.
Another question would be to what degree genomes from the Denisovians or Neanderthals entered the Korean genome. IBD or SNV sharing may be used to assess ancient genome crossing and sharing.
Please notice, the CAMDA challenge is not limited to these questions!
Data download Preprocessed NGS data in VCF format, even merged with data from the 1000 Genomes Project can be downloaded here. Participants who want to use the raw data (BAM-files) should read and accept the data download agreement to get access.








{kind=link}
{kind=link}
{kind=link}
STAY CONNECTED
Tweet